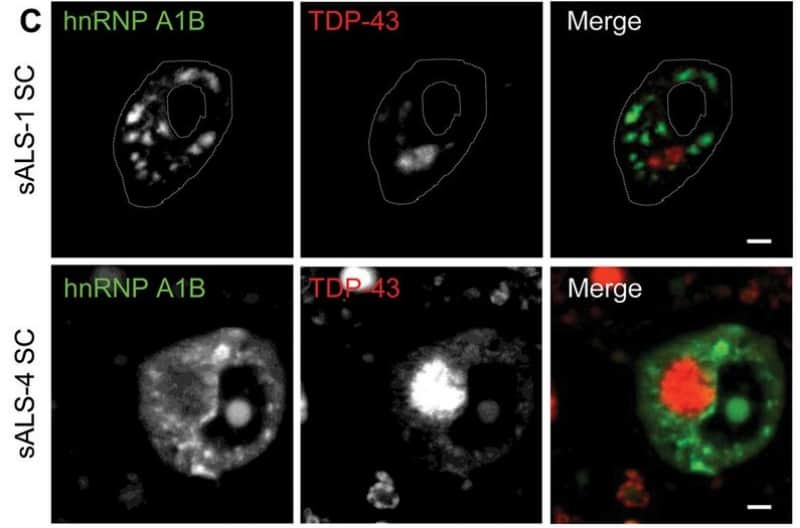
La protéine hnRNP A1B forme des agrégats dans le cytoplasme des cellules des patients atteints de SLA. Photo: CRCHUM.
Une avancée en recherche fondamentale sur la sclérose latérale amyotrophique (SLA) améliore la compréhension du fonctionnement de la maladie.
C’est un parcours de longue haleine: pas moins de 8 ans ont été nécessaires pour décrypter un mécanisme moléculaire impliqué dans la SLA, maladie également connue sous le nom de maladie de Lou Gehrig ou de Charcot, une maladie neurodégénérative incurable. Les personnes atteintes perdent progressivement leurs capacités musculaires jusqu’à la paralysie complète, l’espérance de vie n’étant en moyenne que de 3 à 5 ans après le diagnostic. Le renommé astrophysicien Stephen Hawking en était notamment atteint.
Chez les individus souffrant de SLA, une protéine (TDP-43) est moins présente dans le noyau des cellules, ce qui rend ces dernières plus vulnérables.
«TDP-43 est une protéine ayant plusieurs fonctions dans la régulation de l’ARN qui est ensuite traduit en une autre protéine. Ce sont des composantes essentielles dans le fonctionnement de la cellule», explique Christine Vande Velde, neuroscientifique à l’Université de Montréal, qui fait partie du groupe de chercheurs ayant effectué cette recherche (Hebrew University, Université de Sherbrooke, Western University). Les chercheurs ont examiné que conséquemment à la baisse de cette protéine importante, une autre protéine (hnRNP A1B) a tendance à s’agréger dans le cytoplasme et devient alors toxique pour la cellule. C’est une perturbation cellulaire plus importante que ce que d’autres études avaient constaté.